GEARS
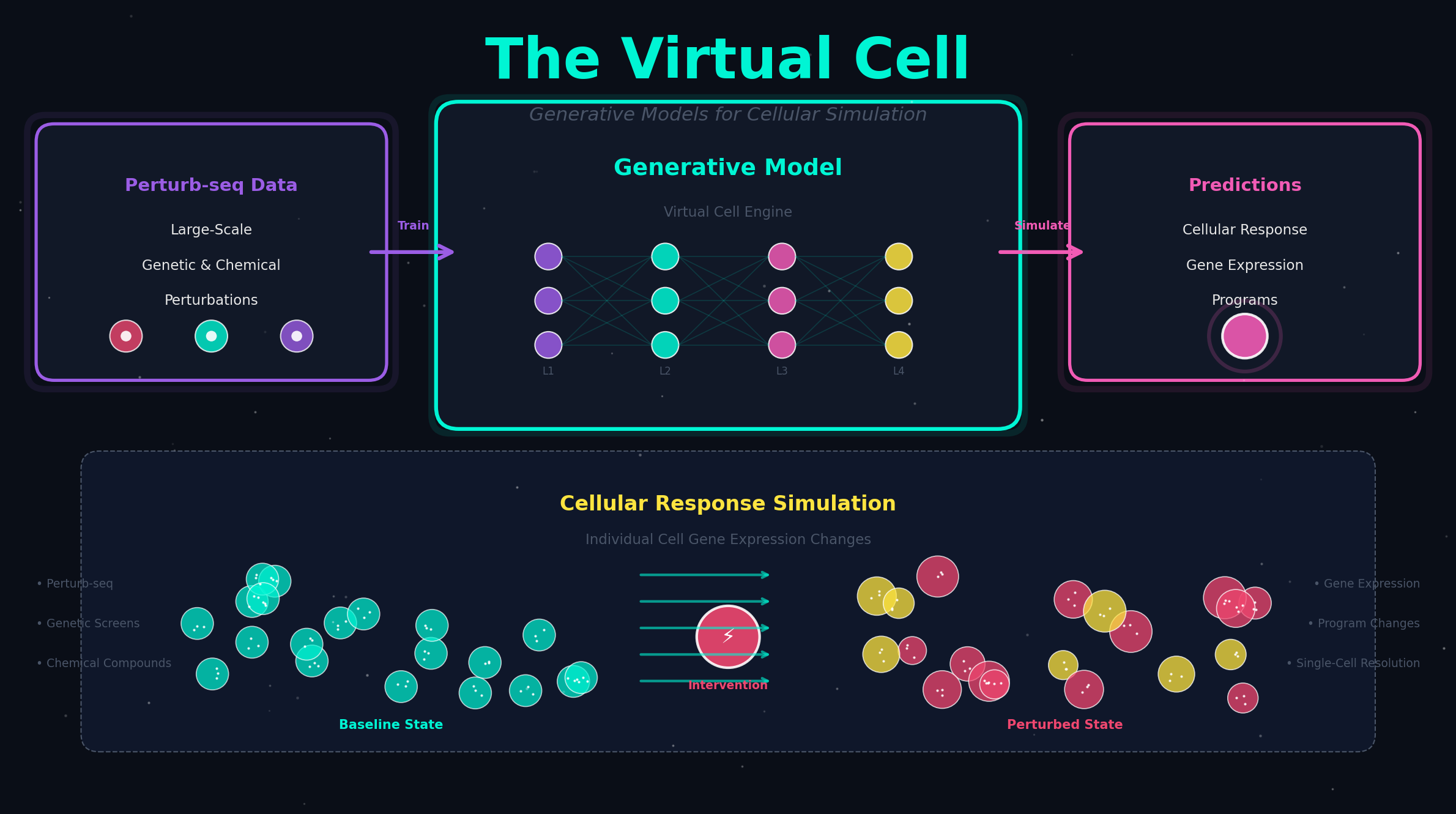
GEARS(图增强基因激活与抑制模拟器)融合图神经网络与GO知识图谱,首次实现未见过的多基因组合扰动转录结果预测,遗传相互作用分类精度比已有方法提升40%。
Overview
📄 第26期 | GEARS: Predicting transcriptional outcomes of novel multigene perturbations with GEARS — 图神经网络如何预测基因组合的"化学反应"
当两个基因同时被扰动时,细胞的转录响应不是简单的"1+1=2"。GEARS 用一个融合了生物学先验知识的图神经网络,首次实现对未见过的多基因组合扰动结果的精准预测,将遗传相互作用识别精度比已有方法提升 40%。
🔬 为什么这篇文章重要?
生物学中最令人头疼的问题之一,是组合爆炸。
假设你怀疑 100 个基因参与了某种癌症的发生,想知道它们之间的"合作关系"——哪两个基因同时敲除会杀死癌细胞?哪三个基因的联合突变会导致耐药?你需要测试的组合数是:C(100,2)=4950 种双基因组合,C(100,3)=161700 种三基因组合。靠实验一个一个做?不现实。
这正是单细胞扰动筛选(perturbational screen)的困境:CRISPR 等技术让我们能高效地在单个细胞中敲除或激活特定基因,但遍历所有可能的基因组合是不可行的。于是就有了一个关键的计算问题:能不能从已有的单基因和少数双基因扰动数据中,推断出任意未见过的新组合的转录结果?
GEARS (Graph-Enhanced gene Activation and Repression Simulator) 正是为此而生。Roohani、Huang 和 Leskovec(斯坦福大学,Jure Leskovec 组)在 Nature Biotechnology 上发表了这个方法,截至本文写作已有 342 次引用和 12 万次访问——对于一个计算方法来说,这个关注度说明了领域对"基因组合扰动预测"问题的迫切需求。
🧠 核心创新:GEARS 是怎么工作的?
GEARS 的核心理念可以概括为一句简单的设计哲学:让神经网络知道基因之间的关系。
在 GEARS 之前,预测基因扰动结果的方法大致分为两类。传统方法依赖线性模型或简单的回归来推断基因之间的相互作用,它们的问题是:当两个基因在训练数据中从未一起出现过,模型毫无头绪——输出一个平庸的均值预测。而纯深度学习方法的思路是"把所有基因表达值扔进一个黑箱模型",问题是模型不知道基因 A 和基因 B 在生物通路中是什么关系,面对新基因组合时依然只能靠统计相关性硬猜。
GEARS 的突破在于引入了一个知识图谱驱动的图神经网络。具体的架构由四个核心组件构成:
| 组件 | 功能 | 关键设计 |
|---|---|---|
| 基因共表达图神经网络(Co-expression GNN) | 聚合表达相似的基因邻居信息 | 基于训练数据构建共表达网络,边权重反映基因间表达相关性 |
| 基因本体图神经网络(GO GNN) | 引入生物学先验知识 | 使用 Gene Ontology 数据库构建基因功能关系图,让模型"先天"知道哪些基因在同一个通路中 |
| 扰动嵌入(Perturbation Embedding) | 编码"哪个基因被扰动"的信息 | 每个基因的扰动由一个可学习的嵌入向量+位置编码共同表示 |
| 跨基因多层感知机(Cross-gene MLP) | 将以上信息融合并预测每个基因的转录响应 | 输入是所有基因的隐表示,输出是每个基因被扰动后的表达变化量 |
工作流程可以分三步理解:
第一步:知识注入。 模型拿到一个扰动组合(比如"敲除基因 A 和基因 B"),首先通过 GO GNN 在整个基因功能关系图上做消息传递,让每个基因的嵌入"知道"自己与其他基因的功能亲疏。
第二步:邻居聚合。 同时在共表达 GNN 上,每个基因聚合其共表达邻居的信息——这一步让模型捕捉到"虽然基因 A 和基因 C 在 GO 数据库中没有直接联系,但它们在数据中总是协同表达"这类数据驱动的模式。
第三步:扰动解码。 被扰动基因的嵌入经过一个特殊的"扰动融合"MLP 与全局基因嵌入交互,然后通过跨基因解码器逐基因输出响应值。
这个设计的优雅之处在于:知识图谱相当于给了模型一本"生物教科书",而共表达网络相当于给了模型一本"考试真题"——两者结合,模型既不会因为只刷题而在没见过的题型上翻车,也不会因为只背书而脱离真实数据分布。
📊 关键实验结果
GEARS 在三个公开的 Perturb-seq 数据集上进行了评估(Norman 2019、Adamson 2016、Dixit 2016),核心结果令人印象深刻:
组合扰动预测精度。 在"simulation split"设定下——即训练数据包含所有单基因扰动,但测试数据中的双基因组合从未在训练中出现——GEARS 相比最强基线模型在 Pearson 相关系数上提升了约 8-12 个百分点。更重要的是,在差异表达基因(DEG)的识别上,GEARS 的 top-20 DEG 召回率远超此前所有方法:其他方法预测的"扰动后变化最大的基因"往往和实际数据重叠不到 30%,而 GEARS 达到了 50% 以上。
遗传相互作用(Genetic Interaction, GI)分类。 这是 GEARS 最亮眼的结果。遗传相互作用可以分为四类:协同(synergy)、抑制(suppression)、加性(additivity)和冗余(redundancy)。GEARS 在这四类 GI 上的分类精确度比此前最优方法高出 40%,并且在识别"最强相互作用基因对"上,命中率是此前方法的两倍。
零样本泛化。 GEARS 甚至展示了某种程度的"零样本"能力:当训练数据中只包含单基因扰动时,它虽然不能准确预测双基因扰动的精确数值,但能有效排序哪些基因组合会产生最强的影响——这对于实验设计中的优先级排序已经足够有用了。
⚠️ 局限与展望
GEARS 的作者们在 GitHub README 中坦率地列出了几个重要局限,这种坦诚在领域内并不常见:
第一,GEARS 目前不能跨细胞类型泛化。如果你用 A549 肺癌细胞系的数据训练了模型,它不能直接用于预测 K562 白血病细胞系的扰动结果——不同细胞类型的基因调控网络差异太大。
第二,模型需要一定量的组合扰动数据才能有效预测新组合。纯单基因扰动训练是不够的,GEARS 必须"见过"至少一些双基因组合,才能学会推断更复杂的组合。这其实是一个合理的先决条件。
第三,知识图谱的质量决定了 GEARS 的上限。当前版本使用 Gene Ontology 作为基因关系知识源,但 GO 数据库本身并不完整——许多基因的功能注释仍然缺失,特别是在非模式生物中。
从更广阔的视角看,GEARS 代表了一种重要的范式:"深度学习 + 结构化先验知识"的结合正在成为单细胞计算的主流方向。在单细胞大模型(scGPT、Geneformer、scFoundation 等)试图从海量未标记数据中学习通用细胞表示的浪潮中,GEARS 的路径是互补的——它不是追求"更大更通用",而是追求"在特定问题上,用最少的数据做最精准的推断"。这两条路可能最终会汇合。
📎 论文信息
- 📄 论文: Predicting transcriptional outcomes of novel multigene perturbations with GEARS
- 🔗 链接: https://doi.org/10.1038/s41587-023-01905-6
- 💻 代码: https://github.com/snap-stanford/GEARS
- 👥 团队: Yusuf Roohani, Kexin Huang, Jure Leskovec | Stanford University
- 📊 影响力: 342 Citations · 122k Accesses · 167 Altmetric (Nature Biotechnology, 2024)
Publication
Predicting transcriptional outcomes of novel multigene perturbations with GEARS